
RESEARCH
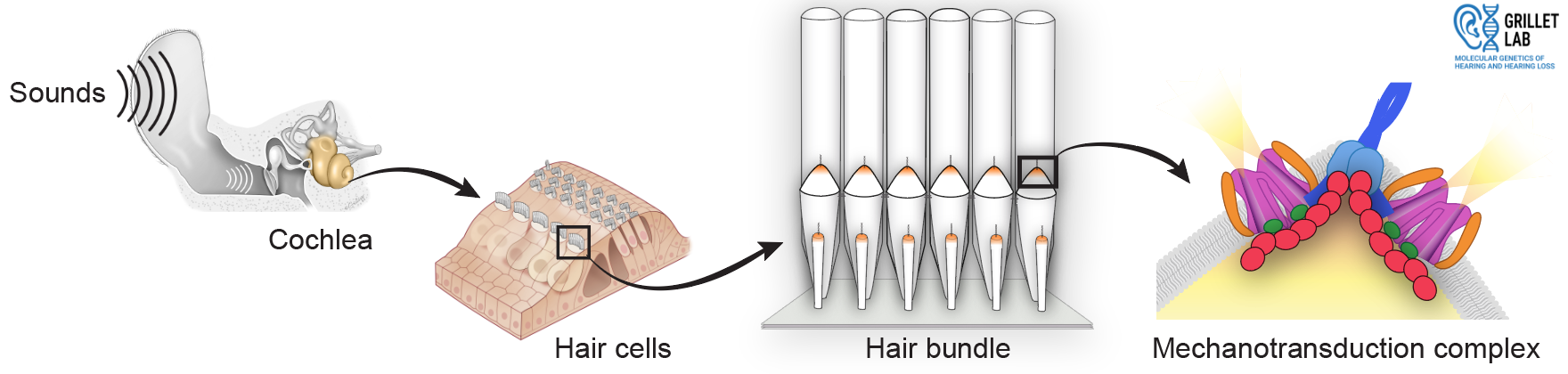
Hearing loss affects 20% of people worldwide and is often caused by problems with inner ear sensory cells called hair cells.
Hair cells detect sound-induced vibrations and convert them into electrical signals that the brain can understand.
In mammals, hair cells form before birth and do not regenerate, so it's essential to understand how hair cells work and how to fix them when they stop working.
ONGOING PROJECTS
Molecular Mechanisms of Hearing
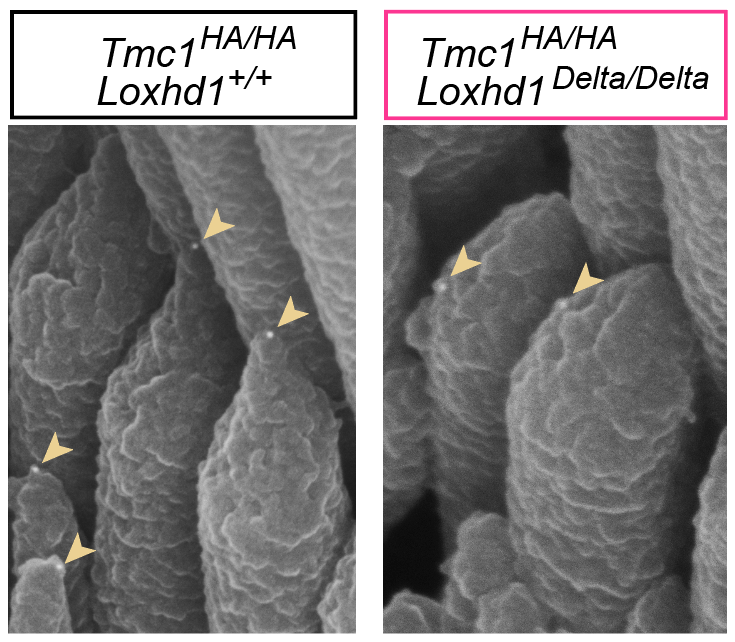
We identified LOXHD1 as the gene responsible for the recessive form of hearing loss DFNB77 (Grillet et al., AJHG, 2009). In mouse models, we showed that Loxhd1 mutations impair auditory mechanotransduction without disrupting tip links or hair bundle morphology (Trouillet et al., J. Neurosci., 2021). More recently, we demonstrated that loss of LOXHD1 leads to the progressive mislocalization of TMC1 channels from their normal position at the stereocilia tips, preventing their mechanical activation by sound (Wang et al., Nat. Commun., 2024). In the same study, we also showed that LOXHD1 can bind, in vitro, to several other components of the mechanotransduction complex. Our current goal is to map these protein interactions in detail and determine whether LOXHD1 influences the biophysical properties of the auditory transduction channels.
Molecular Genetics of Human Age-Related Hearing Loss
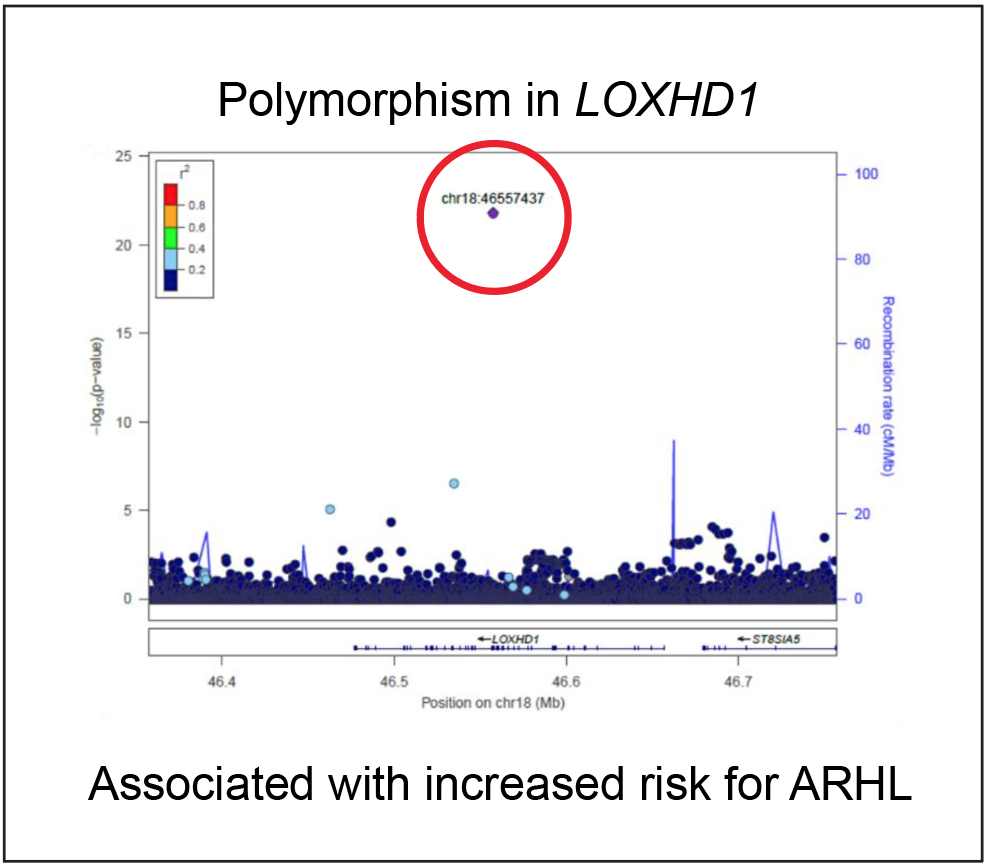
Gradual hearing loss is a common consequence of aging. Age-related hearing loss (ARHL) is a complex neurosensory disorder influenced by both genetic and environmental factors. Until recently, the genetic basis of ARHL was poorly understood. A large-scale genome-wide association study (GWAS) has now identified 51 genetic variants associated with ARHL, highlighting the complexity of its genetic architecture (Ivarsdottir et al., Commun. Bio., 2021). Despite this advance, the causal role of these variants and their specific effects on the auditory system remain largely uncharacterized.
Our central hypothesis is that ARHL-associated variants and genes operate within interconnected molecular networks. In this project, we aim to define one such network, beginning with a variant in LOXHD1, a gene we have studied extensively.
Given the high evolutionary conservation of hearing-related genes and mechanisms across mammals, we propose that the ARHL-associated missense variant in LOXHD1 can be functionally modeled in mice to evaluate its pathogenicity. We have generated this mouse model and will determine whether the variant is indeed causal. In addition to longitudinal hearing assessments, we will investigate whether the polymorphism affects susceptibility to noise-induced hearing loss.

Therapeutic Approach to Hearing Loss
We have developed a series of mouse models carrying Loxhd1 dysfunction, which allows us to investigate the functional requirements of this gene in auditory physiology. Our objective is to determine which Loxhd1 splice variant(s) are necessary and sufficient to preserve hearing function. To address this question, we are employing a combination of genetic approaches, including isoform-specific rescue using knock-in strategies, alongside gene therapy techniques such as adeno-associated virus (AAV)-mediated delivery of individual splice variants. This integrated approach will enable us to dissect the specific contributions of each Loxhd1 isoform to auditory function and guide the development of targeted therapeutic strategies for LOXHD1-related hearing loss.